


科研與服務 SERCIVE
聯系我們 CONTACT US
第四節 影響PCR的主要因素
PCR技術必須有人工合成的合理引物和提取的樣品DNA,然后才進行自動熱循環,最后進行產物鑒定與分析。引物設計與合成目前只能在少數技術力量較強的研究院、所進行,臨床應用只需購買PCR檢測試劑盒就可開展工作,PCR自動熱循環中影響因素很多,對不同的DNA樣品,PCR反應中各種成份加入量和溫度循環參數均不一致。現將幾種主要影響因素介紹如下。
一、溫度循環參數
在PCR自動熱循環中,最關鍵的因素是變性與退火的溫度。如操作范例所示,其變性、退火、延伸的條件是:94℃60s, 37℃60s, 72℃120s,共25~30個循環,擴增片段500bp。在這里,每一步的時間應從反應混合液達到所要求的溫度后開始計算。在自動熱循環儀內由混合液原溫度變至所要求溫度的時間需要30~60s,這一遲滯時間的長短取決于幾個因素,包括反應管類型、壁厚、反應混合液體積、熱源(水浴或加熱塊)以及兩步驟間的溫度差,在設置熱循環時應充分給以重視和考慮,對每一儀器均應進行實測。
關于熱循環時間的另一個重要考慮是兩條引物之間的距離;距離越遠,合成靶序列全長所需的時間也越長,前文給出的反應時間是按最適于合成長度500bp的靶序列擬定的。下面就各種溫度的選擇作一介紹。
1.模板變性溫度變性溫度是決定PCR反應中雙鏈DNA解鏈的溫度,達不到變性溫度就不會產生單鏈DNA模板,PCR也就不會啟動。變性溫度低則變性不完全,DNA雙鏈會很快復性,因而減少產量。一般取90~95℃。樣品一旦到達此溫度宜迅速冷卻到退火溫度。DNA變性只需要幾秒種,時間過久沒有必要;反之,在高溫時間應盡量縮短,以保持Taq DNA聚合酶的活力,加入Taq DNA聚合酶后最高變性溫度不宜超過95℃。
2.引物退火溫度退火溫度決定PCR特異性與產量;溫度高特異性強,但過高則引物不能與模板牢固結合,DNA擴增效率下降;溫度低產量高,但過低可造成引物與模板錯配,非特異性產物增加。一般先由37℃反應條件開始,設置一系列對照反應,以確定某一特定反應的最適退火溫度。也可根據引物的(G+C)%含量進行推測,把握試驗的起始點,一般試驗中退火溫度Ta(annealing temperature)比擴增引物的融解溫度TTm(melting temperature)低5℃,可按公式進行計算:
Ta = Tm - 5℃= 4(G+C)+ 2(A+T) -5℃
其中A,T,G,C分別表示相應堿基的個數。例如,20個堿基的引物,如果(G+C)%含量為50%時,則Ta的起點可設在55℃。在典型的引物濃度時(如0.2μmol/L),退火反應數秒即可完成,長時間退火沒有必要。
3.引物延伸溫度溫度的選擇取決于Taq DNA聚合酶的最適溫度。一般取70~75℃,在72℃時酶催化核苷酸的標準速率可達35~100個核苷酸/秒。每分鐘可延伸1kb的長度,其速度取決于緩沖溶液的組成、pH值、鹽濃度與DNA模板的性質。擴增片段如短于150bp,則可省略延伸這一步,而成為雙溫循環,因Taq DNA聚合酶在退火溫度下足以完成短序列的合成。對于100~300bp之間的短序列片段,采用快速、簡便的雙溫循環是行之有效的。此時,引物延伸溫度與退火溫度相同。對于1kb以上的DNA片段,可根據片段長度將延伸時間控制在1~7min,與此同時,在PCR緩沖液中需加入明膠或BSA試劑,使Taq DNA聚合酶在長時間內保持良好的活性與穩定性;15%~20%的甘油有助于擴增2.5kb左右或較長DNA片段。
4.循環次數常規PCR一般為25~40個周期。一般的錯誤是循環次數過多,非特異性背景嚴重,復雜度增加。當然循環反應的次數太少,則產率偏低。所以,在保證產物得率前提下,應盡量減少循環次數。
擴增結束后,樣品冷卻并置4℃保存。
二、引物引物設計
要擴增模板DNA,首先要設計兩條寡核苷酸引物,所謂引物,實際上就是兩段與待擴增靶DNA序列互補的寡核苷酸片段,兩引物間距離決定擴增片段的長度,兩引物的5’端決定擴增產物的兩個5’末端位置。由此可見,引物是決定PCR擴增片段長度、位置和結果的關鍵,引物設計也就更為重要。
引物設計的必要條件是與引物互補的靶DNA序列必須是已知的,兩引物之間的序列未必清楚,這兩段已知序列一般為15~20個堿基,可以用DNA合成儀合成與其對應互補的二條引物,除此之外,引物設計一般遵循的原則包括:
1.引物長度根據統計學計算,長約17個堿基的寡核苷酸序列在人的基因組中可能出現的機率的為1次。因此,引物長度一般最低不少于16個核苷酸,而最高不超過30個核苷酸,最佳長度為20~24個核苷酸。這樣短的寡核苷酸在聚合反應溫度(通過72℃)下不會形成穩定的雜合體。有時可在5’端添加不與模板互補的序列,如限制性酶切位點或啟動因子等,以完成基因克隆和其他特殊需要;引物5’端生物素標記或熒光標記可用于微生物檢測等各種目的。
有時引物不起作用,理由不明,可移動位置來解決。
2.(G+C)%含量引物的組成應均勻,盡量避免含有相同的堿基多聚體。兩個引物中(G+C)%含量應盡量相似,在已知擴增片段(G+C)%含量時宜接近于待擴增片段,一般以40%~60%為佳。
3.引物內部應避免內部形成明顯的次級結構,尤其是發夾結構(hairpin structures)。
例如:
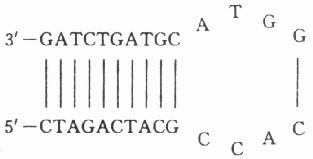
4.引物之間兩個引物之間不應發生互補,特別是在引物3’端,即使無法避免,其3’端互補堿基也不應大于2個堿基,否則易生成“引物二聚體”或“引物二倍體”(Primer dimer)。所謂引物二聚體實質上是在DNA聚合酶作用下,一條引物在另一條引物序列上進行延伸所形成的與二條引物長度相近的雙鏈DNA片段,是PCR常見的副產品,有時甚至成為主要產物。
另外,兩條引物之間避免有同源序列,尤為連續6個以上相同堿基的寡核苷酸片段,否則兩條引物會相互競爭模板的同一位點;同樣,引物與待擴增靶DNA或樣品DNA的其它序列也不能存在6個以上堿基的同源序列。否則,引物就會與其它位點結合,使特異擴增減少,非特異擴增增加。
5.引物3’端配對DNA聚合酶是在引物3’端添加單核苷酸,所以,引物3’端5~6個堿基與靶DNA的配對要求必須精確和嚴格,這樣才能保證PCR有效擴增。
引物設計是否合理可用PCRDESN軟件和美國PRIMER軟件進行計算機檢索來核定。
人工合成的寡核苷酸引于最好經過色譜(層析)純化或PAGE純化,以除去未能合成至全長的短鏈等雜質。純化引物在25%乙腈溶液中4℃保存可阻止微生物的生長;一般情況下,不用的引物應保存在-20℃冰箱中,在液體中引物能保存6個月,凍干后可保存1~2年。
三、DNA聚合酶
早在1956年Kornberg等就從大腸桿菌提取液中發現了DNA聚合酶,并且得到了DNA聚合酶Ⅰ純品。DNA聚合酶Ⅰ是由分子量為109000的一條多肽鏈構成,此酶可被枯草桿菌蛋白酶分解為兩個片段,一個片段分子量為76000,有聚合酶活性,并有3’→5外切酶活力,即Klenow片段(Klenow fragment)。另一個片段分子量為34000,具有5’→’3’外切酶活力。因此,DNA聚合酶具有幾種功能:一是聚合作用,以DNA為模板,將dNTP中的脫氧單核苷酸逐個加到3-OH末端。二是有’3’→5’外切酶活力,能識別和消除錯配的引物末端,與復制過程中校正功能有關。三是5’→3’外切酶活力,它能從5’端水解核苷酸,還能經過幾個核苷酸起作用,切除錯配的核苷酸。1985年Mullis 等發明了PCR方法,以Klenow片段完成β-珠蛋白的PCR后,世界上許多實驗室就考慮用耐熱DNA聚合酶代替Klenow片段進行PCR,使耐熱多聚酶的研究得以迅速發展。人們從生活于60℃(B.Stearothermophilus)到87℃(S.Solfatavicus)的許多菌中分離純化出耐熱DNA聚合酶,但有些酶不能耐受DNA變性所需溫度,所以無法應用于PCR。現就PCR反應中常用的DNA聚合酶等作一詳細介紹。
1.Taq DNA聚合酶用Taq DNA聚合酶代替大腸桿菌DNA聚合酶Ⅰ的Klenow片段是使PCR普及應用的關鍵。Klenow片段不能耐受95℃的雙鏈DNA變性溫度,所以每次循環都要加入新酶;而Taq DNA聚合酶可以耐受93~95℃的高溫,避免了不斷補加多聚酶的繁瑣操作,同時使退火和延伸溫度得以提高,減少了非特異性產物和DNA二級結構對PCR的干擾,增進了PCR特異性、產量和敏感度,二者相比,其主要區別在于:①Klenow酶的最適溫度為37℃,擴增的產物并非全是目的序列,需用探針檢測。Taq酶則不僅產率高而特異性也高。它的最適溫度為74~75℃。因而使退火溫度可以提高,使退火嚴格性提高,減少錯配引物的延伸。②循環后期酶量漸感不足而產生平坡。到達平玻的循環次數,Klenow酶為20個(均用1μg基因組DNA開始)而Taq酶為30個。③延伸片段長度Taq酶為10kb以內,而Klenow酶為400bp以內。
Taq酶由水棲高溫菌(Thermus aquatics)YT1蓖株中分離而得。此菌于1969年由Brock分離自美國黃石公園溫泉,作為棲熱桿菌的標準菌株,其生長溫度為70~75℃。最初從中分離到分子量60~68KDa,比活性為2000~8000U/mg的DNA聚合酶。后來Cetus公司的Kary Mullis等又分離到比活為20萬U/mg的純酶,分子量為93910。此種9.4KDa酶的最適溫度為75~80℃,與單純核苷酸的結合率(Kcat)可達150核苷酸(nt)/s酶分子。以M13模板,用富含G+C的30bp引物延伸,70℃時Kact>60nt/s;55℃可達24nt/s;37℃時為1.5nt/s,而22℃時低至0.25nt/s。高于90℃時DNA合成活性甚差,這種高溫條件下,引物與模板已不能牢固結合。
在PCR反應混合液中,Taq酶于92.5℃,95℃及97.5℃保持其50%活力的時間分別為130、40及5~6min,在50次循環的PCR中當管內最高溫度為95℃。每循環為20s時尚可保持65%活力。Taq 酶在95℃的半壽期為40min,故在PCR循環中選用的變性溫度,不宜高于95℃。
Taq酶現已可用基因重組的方法生產,商品名為Ampli Taq(Cetus公司)。Taq酶的完整基因長2499bp,在大腸桿菌中表達生產,含832個氨基酸。在氨基酸序列上與大腸桿菌DNA聚合酶Ⅰ有38%是一致的,包括對dNTP結合,引物與模板作用區均存在于Taq酶中。
Taq酶具有依賴DNA合成的5’→’3’外切酶活性,因此,模板上有一段退火的3’-磷酸化的“阻斷物”,會被逐個切除而不會阻止來自上游引物鏈的延伸,而對于5’-32P標記的合成寡核苷酸引物,則無論是單鏈或是與模板復性,都未發現降解,所以該種活性不會影響PCR結果。Taq酶沒有3’→’5’外切酶活性,如果發生dNTP錯誤摻入,這種酶沒有校正能力,因此運用Taq酶進行PCR,產物中點突變較多,對克隆等不太有利。一般錯摻率為1.25×10-4~1×10-5(4×dNTPs濃度分別為200μmol/L,Mg2+為1.5mmol/L,在55℃退火)。但不含3’→5’外切酶活性對測序有利。
2.影響酶活力的因素Taq酶的活力受Mg2+離子的影響。用鯡精DNA為模板,總dNTP濃度0.7~0.8mmol/L,Mg2+為2.0mmol/L時激活能力最高。濃度超過此值產生抑制。10mmol/l MgCl2抑制活力達40%~50%。dNTP能與Mg2+結合,故游離Mg2+只是結合后剩余的量。若總dNTP濃度高至4~6mmol/L時,Taq酶活力要降低20~30%,即底物抑制。
dNTP濃度低時PCR產率及特異性均增高,適合于用擴增摻入法標記生物素及放射性元素。當100μl PCR液中含dNTP各40μmol/L時就足以合成2.6μg的DNA(dNTP消耗一半)。
表22-3 有機溶劑對Taq聚合酶活力的影響
用鯡精DNA,70℃,10min內dNTP的摻入量計算,標準條件為100%。
純9.4KDa Taq酶不含3’→5’核酸外切酶活力。誤摻入率取決于dNTP濃度。但Taq酶具有DNA依賴的鏈移位5’→3’核酸外切酶活力。對5’→3’32P標記寡核苷酸單鏈,或與MB模板雜交時均只有極少的降解力。
中等濃度KCl能刺激Taq酶合成活力達50%~60%,最佳KCl濃度為50mmol/L,濃度更高有抑制作用,>200mmol/L的KCl可使酶失活。
加入50mmol/L NH4Cl或NH4Ac或NaCl,可產生中度抑制或無作用。
低濃度尿素、DMSO、DMF或甲酰胺影響不大,吐溫20/NP40可消除SDS(0.01%及0.1%)的抑制作用。
3.第二代耐熱DNA聚合酶Stoffel片段:Cetus公司的Stoffel將Taq DNA聚合酶的5’→3’外切酶活性片段(N端289個氨基酸)去除,稱為stoffel片段。其97.5℃的半衰期從Taq DNA聚合酶的5~6min提高到20min,同時該酶片段也對兩個或更多模板位點的擴增反應即復合PCR(Multiplex PCR)更為有利。
VentTM DNA多聚酶:是美國New England Biolabs公司從潛水艇排氣孔(Vent)中分離的超級嗜熱菌-能生長于98℃中的Thermococcus litoralis中分離純化得到的,故名Vent酶。它的一些酶學性質較Taq DNA聚合酶更為優越,它能耐100℃高溫且2h以上仍有活力,并且具有3’→5’外切酶活性的校正能力,錯誤擴增的機率比Taq酶降低一倍。后來該公司又從深水潛艇(2010m)排氣孔分離的能在104℃生長的Pyococcus菌GB-D株植入Deep Vent DNA聚合酶基因而表達的Deep Vent DNA聚合酶,在95℃的半壽期達23h(Vent酶為6.7h,Taq酶為1h)。
4.RTth逆轉錄酶(rTth Reverse Transcriptase)目前逆轉錄-PCR(RT-PCR)的發展很快,所以對耐熱的依賴于RNA的DNA多聚酶的研究也有進展。有實驗表明Taq DNA多聚酶有依賴于RNA的DNA聚合酶活性,但活性較弱。Cetus公司于1991年推出一種rTth Reverse Tran-scriptase,有很好的依賴于RNA的耐熱DNA聚合酶活性和依賴于DNA的耐熱DNA聚合酶活性,二種活性分別依賴于Mn2+Mg2+,這樣就可分別控制酶活性。利用該酶只需250ng的總RNA即可有效地進行RT-PCR,得到特異的DNA片段,從而非常有利于逆轉錄PCR的發展。
耐熱DNA聚合酶的研究近幾年來得到長足的發展,這在PCR發展中起到了重要的作用。我們相信隨著進一步的研究,將使人們對耐熱DNA聚合酶的認識和應用更進一步地發展。
我國的PCR研究發展很快,其關鍵試劑-耐熱DNA聚合酶-也已有幾個實驗室能夠分離純化,如復旦大學遺傳學研究所、華美公司、中國醫學科學院基礎醫學研究所。后二者的菌株為Thermus aquaticus YT-1。前者則是從自己篩選的嗜熱菌中分離純化,復旦大學遺傳所亦已成功地克隆了該聚合酶的基因并獲得了耐熱F4DNA聚合酶,其酶學性質非常接近于Taq DNA聚合酶,為我國PCR的開展提供了保證。
四、影響PCR特異性的因素
通過上述內容。可以看出有許多因素可以影響PCR的特異性,在此我們作一歸納,供大家參考:①退火步驟的嚴格性:提高退火溫度可以減少不匹配的雜交,從而提高特異性。②減短退火時間及延伸時間可以減少錯誤引發及錯誤延伸。③引物二聚體是最常見的副產品,降低引物及酶的濃度也可以減少錯誤引發,尤其是引物的二聚化。④改變MgCl2(有時KCl)濃度可以改進特異性,這可能是提高反應嚴格性或者對Taq酶的直接作用。⑤模板中如果存在次級結構,例如待擴增的片段易自行形成發夾結構時,可在PCR混合物中的4×dNTPs中加入7-脫氮-2’-脫氧鳥苷-5’-三磷酸(7-deaza-2’-deoxyguanosine-5’-trihosphate)(de7GTP)。用de7GTP與dGTP比例為3:1的混合物(150μmol/l de7GTP +50μmol/L dGTP)代替200μmol/l dGTP,則可阻非特異性產物的生成。
五、擴增平坡
擴增反應并不是可以無窮地進行下去的,經過一定的循環周期后需擴增的片段不再按指數增多而逐漸進入平坡;進入平坡的循環次數,取決于起始時存在的模板拷貝數以及合成的DNA總量。所謂平坡就是批PCR循環的后期,合成產物達0.3~1pmol時,由于產物的堆積,使原來以指數增加的速率變成平坦的曲線。
造成PCR進入平坡的原因有:引物和dNTP等消耗完畢、Taq酶失活,這幾中因素在標準反應中均不會出現。此外,還有幾種可能:
1.底物過剩 因DNA合成量多于反應液中存在的Taq酶,在100μl反應液中含2.5Utaq酶而DNA合成量達1μg(3nmol脫氧核苷酸)時,開始變為底物過剩。延長延伸時間或添加Taq酶,可以克服之。但不實用,因每進行下一循環就要延長延伸時間一倍及多加一倍Taq酶,才能繼續保持指數增長。
2.非特異性擴增產物的競爭 與上述情況密切相關,此時不需要的DNA片段與需要的片段同時競爭聚合酶,要克服這一情況是要提高反應特異性,使不需要片段不能大量積聚。
3.退火時產物的單鏈自己締合 兩條單鏈的DNA片段在退火時除了與引物締合外,也可以自行締合,這也會阻止產品增多。當產物濃度到達10pmol/100μl時即可發生此現象,除稀釋外無法克服。
4.變性在高濃度產物條件下,產物解鏈不完全,以及最終產物的阻化作用(焦磷酸化,雙鏈DNA)。
總而言之,PCR的條件是隨系統的而異的,并無統一的最佳條件,先選用通用的條件擴增,然后稍稍改變各參數,可以達到優化,以取得優良的特異性和產率。
合作伙伴/PARTNER
版權所有:北京金諾美科技股份有限公司